INTRODUCTION
Gorlin–Goltz syndrome, which is also known as nevoid basal cell carcinoma (BCC) syndrome, was first reported by Jarish and White in 1894.1 It is a rare autosomal dominant disorder with strong penetrance and extremely variable expressivity. Gorlin and Goltz2 described the distinct syndrome, consisting of multiple nevoid BCCs, jaw cysts, and bifid ribs. A spectrum of other neurological, ophthalmic, endocrine, and genital manifestations is known to be variably associated with this triad. The incidence of this syndrome is estimated to be 1 in 50,000 to 1,50,000 in the general population, but perceived incidence may vary by region. Males and females are equally affected.3 The pathogenesis of Gorlin–Goltz syndrome is attributed to abnormalities linked to the long arm of chromosome 9 (q22.3-q31). It has been reported that loss of human patched gene (PTCH1 gene), which is a tumor suppressor gene, could be the molecular origin of the syndrome.4 This syndrome is also named as Gorlin syndrome, nevoid BCC syndrome, basal cell nevus syndrome, multiple basalioma syndrome, fifth phacomatosis, hereditary cutaneomandibular polyoncosis, multiple nevoid basal cell epithelioma, jaw cyst bifid rib syndrome, or multiple nevoid BCC syndrome.5
CASE REPORT
Case 1
A 14-year-old male patient first reported to the Department of Oral and Maxillofacial Surgery, Gian Sagar Dental College & Hospital, India, with the chief complaint of swelling in the right lower back region of the jaw for last 2 months. Swelling increased gradually in size over a period of 2 months and was not associated with any pain or discharge. On intraoral examination, swelling was soft, fluctuant, and nontender on palpation. On extraoral examination, the face appeared bilaterally symmetrical, although frontal bossing was revealed along with hypertelorism, broad nasal bridge, relative macrocephaly (Figs 1A to D). Orthopantomograph revealed multiple well-defined radiolucencies with sclerotic borders in the mandible and maxilla (Figs 2A to C). The presence of multiple cysts in the jaws and extraoral examination raised a suspicion of Gorlin–Goltz syndrome, so other relevant clinical examination and investigations were done. On examination of the hands and feet, palmar and plantar pitting was noticed. Multiple nevi were also seen (Figs 1A to D). Posteroanterior skull revealed calcification of falx cerebri (Fig. 3). Radiograph of chest appeared normal.
Surgical enucleation of the cyst was done followed by peripheral ostectomy and chemical cauterization (Figs 4A to F). The sample obtained was then sent for histopathological examination, which revealed parakeratinized stratified squamous epithelium with a palisading pattern of columnar cells along with keratin flakes, suggestive of odontogenic keratocyst.
Based on the clinical, radiographic, and histologic findings, the patient was diagnosed as having Gorlin–Goltz syndrome. The patient is being followed up for last 8 years, at intervals of every 6 months and the required definitive treatment is being instituted to the patient (Figs 4A to F).
Figs 1A to D
(A) Frontal bossing and hypertelorism; (B) palmar pitting; (C) relative macrocephaly; and (D) multiple nevi


Case 2
Another patient, a 20-year-old male, first reported to our department with the chief complaint of swelling in the right lower back region of jaw for last 1 month. Swelling increased gradually in size and was not associated with pain or any discharge. On intraoral examination, swelling was soft, fluctuant and nontender on palpation. On extraoral examination, face showed frontal bossing, hypertelorism, wide nasal bridge, and multiple nevi (Figs 5A to D). Orthopantomogram was done taking into consideration the chief complaint of the patient and it revealed multiple well-defined radiolucencies in the mandible (Figs 6A and B). Taking into account all the above features, other relevant examination and investigations were conducted. On examination of other parts of the body, palmar and plantar pitting was noticed. Multiple nevi were also seen on different parts of the body (Figs 5A to D). Sections of computed tomography head revealed calcification of falx cerebri (Fig. 7).
Figs 4A to F
Surgical enucleation of cyst and peripheral ostectomy and chemical cauterization: (A) Rt mand angle region at age 14; (B) Specimen with extracted Rt mand 3rd molar; (C) Specimen with extracted Rt mand 3rd premolar (D) Rt mand body region at age 17; (E) Rt max antrum region at age 21; and (F) Specimen with extracted Rt max 3rd molar

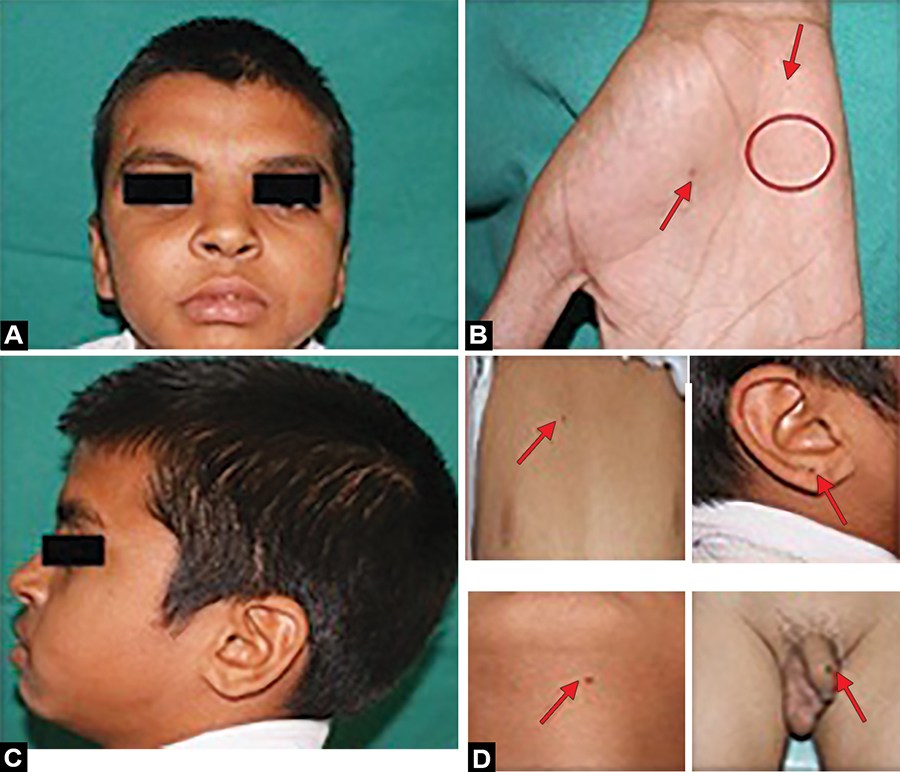
Figs 5A to D
(A) Frontal bossing and multiple nevi; (B) hypertelorism; (C) multiple nevi; and (D) plantar pitting

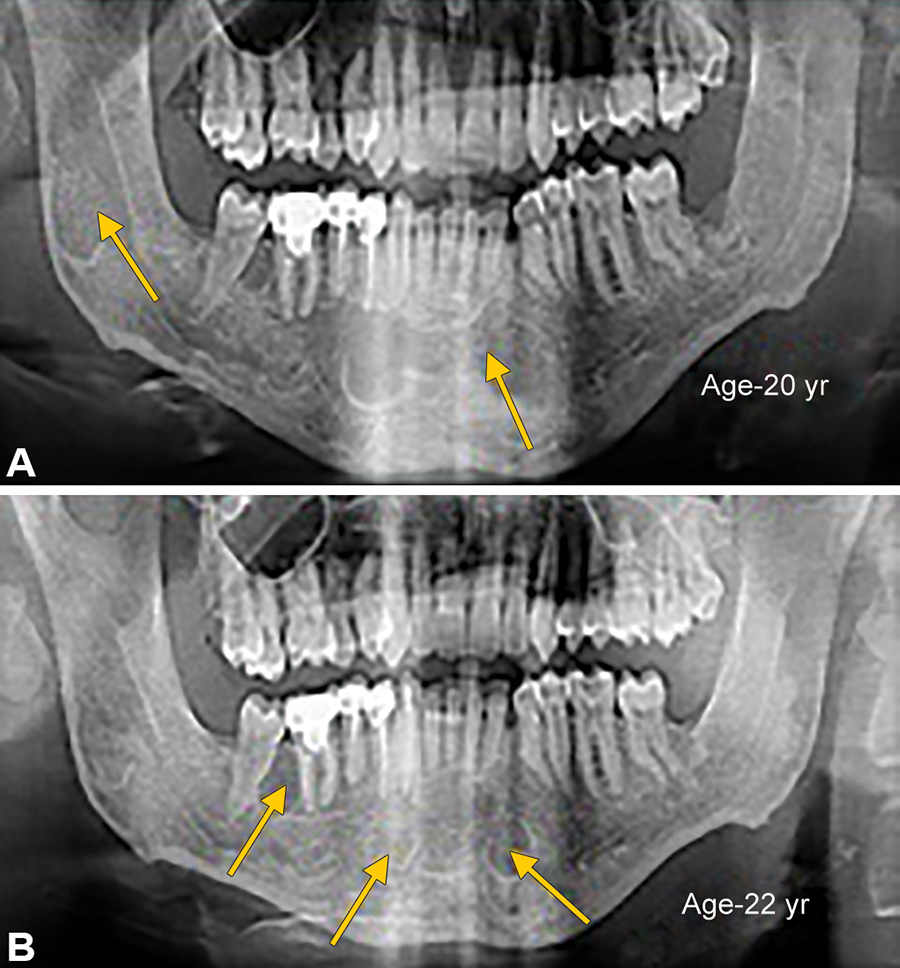
Figs 6A and B
(A) Cystic lesion in Rt ramus region and mand anterior region; (B) cystic lesion in Rt ramus region and recurrent cystic lesion in mand anterior region

The same treatment was instituted as was done in our first patient. Surgical enucleation of the cysts was done followed by peripheral ostectomy and chemical cauterization (Figs 8A to C). The histopathological report was suggestive of the features of odontogenic keratocyst. So, based on the clinical, radiological, and histopathological features, a definitive diagnosis of Gorlin–Goltz syndrome was made. The patient is being followed up for last 3 years at regular intervals of 6 months.

DISCUSSION
Gorlin–Goltz syndrome existed during dynastic Egyptian times, as shown by findings compatible with the syndrome in mummies dating back to 1000 bc.6 In 1894, Jarish and White made the first descriptions of patients with this syndrome.
Gorlin–Goltz syndrome is an autosomal dominant inherited syndrome manifested by multiple defects involving the skin, nervous system, eyes, endocrine system, and bones. It is also known as basal cell nevus syndrome, multiple BCC syndrome, Gorlin syndrome, or hereditary cutaneomandibular polyoncosis, multiple nevoid basal cell epithelioma – jaw cysts, or bifid rib syndrome.5
Globally, the estimated prevalence is from 1 in 50,000 to 1 in 1,50,000 and is equally reported in males and females.7,8 There is no sexual predilection. Individuals with no known affected family members may comprise up to 60% of all affected individuals.9 There are many dermatological, neurological, vertebral, ocular, genitourinary disorders which may be associated with this syndrome.10,11 Together with these major factors, more than 100 minor features have been described.
Evans et al12 first established major and minor criteria for the diagnosis of this rare entity, later modified by Kimonis et al.13 Diagnosis of Gorlin–Goltz syndrome can be established on the basis of the presence of two major criteria or one major and two minor criteria.
Minor Criteria
Macrocephaly
Congenital anomalies (cleft lip or palate, frontal bossing, coarse facies, and moderate or severe hypertelorism)
Other skeletal anomalies (Sprengel deformity, marked pectus deformity, and marked syndactyly of the digits)
Radiological abnormalities (such as bridging of the sella turcica, vertebral anomalies, modeling defects of the hand and feet, or flame-shaped lucencies of hand and feet)
Ovarian fibroma
Medulloblastoma
Several diagnostic modalities can be used for the detection and investigation of Gorlin–Goltz syndrome. Diagnostic protocol accounts for thorough clinical examination that includes an examination of oral cavity, skin, central nervous system, head circumference, interpupillary distance, eyes, genitourinary system, cardiovascular system, respiratory system, and skeletal system. Complete family history including past medical history and dental history should be taken. Genetic testing is also recommended. The radiological examination comprises radiographs of the chest, anteroposterior (AP) and lateral skull view, panoramic radiograph, cervical and thoracic spine – AP and lateral view. Ovarian ultrasound is done in the case of females to rule out ovarian fibroma, and echocardiogram is done in children to rule out cardiac fibroma.14 The investigations prompt an early verification of the disease, which is important to prevent recurrence and better survival rates.
Differential diagnosis should be done mainly from a few rare dermatological disorders, like Bazex syndrome, trichoepithelioma papulosum multiplex, and Muir–Torre's syndrome.15
Various treatment modalities have been described in the literature for the treatment of odontogenic keratocysts. These are marsupialization or decompression (Partsch I procedure), Waldron's operation, enucleation with peripheral ostectomy, chemical treatment with Carnoy's solution, physical treatment with cryotherapy, and resection.16 Excision of the overlying attached mucosa has also been suggested for the elimination of epithelial islands and microcysts located in the mucosa.17
Taking into consideration the age of both the patients and the desirability of esthetics, a more conservative treatment option was chosen in our cases. Surgical enucleation followed by peripheral ostectomy and chemical cauterization were done in both the cases, which provided satisfactory results.
CONCLUSION
To summarize, it can be said that Gorlin–Goltz syndrome is a dominant autosomal disorder, which is of particular interest to an oral and maxillofacial surgeon. A thorough clinical examination and investigations prompts an early verification of the disease, which is important to prevent recurrence and for better survival rates for the existent disease. An odontogenic keratocyst of the jaws are the first manifestations of this syndrome and can cause disfigurement of the face, mobility, and even loss of teeth that can be avoided by early detection and treatment of the same. Periodic follow-ups should be offered in such patients to increase the chances for better overall survival rates.